

Edited by Matteo Iafrate - Pharmaceutical Analytical Chemist
Quantum mechanics allows us to obtain very accurate results in the study of molecules. Over the years, numerous computational methods have been developed that are capable of studying the system under examination with different approaches, always rigorously based on quantum theories. Quantum mechanics attempts to interpret, with excellent results, through statistical probability, the behavior of matter and its energy at the atomic and subatomic level, differentiating itself from classical mechanics, which applies to macroscopic systems. It is important to underline that classical mechanics fails at the molecular level because several properties such as electron delocalization and chemical bonding are strictly linked to quantum effects. Furthermore, quantum mechanical methods offer high accuracy for small systems, while classical methods can handle large systems with reduced accuracy.
Theoretical Foundations
Some of the main theoretical aspects of quantum theory concern wave-particle duality, the quantization of energy, wave function and probability, the superposition principle, the Heisenberg uncertainty principle, and entanglement. The study of time evolution, which describes how the wave function changes over time, with a probabilistic approach, is described by the Schrödinger equation:
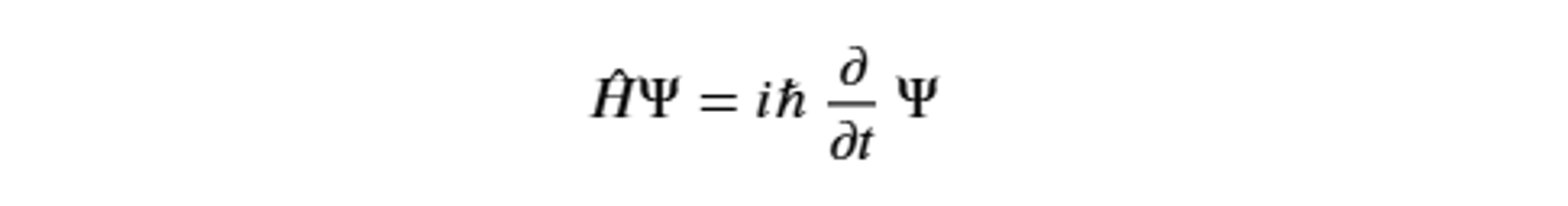
In scientific literature, the use of computational methods that make use of quantum mechanics is increasingly present. Among the most widely used are: density functional theory (DFT), Hartree-Fock (HF), quantum mechanics/molecular mechanics (QM/MM), and fragment molecular orbitals (FMO).
Methods and applications
These techniques are based on quantum mechanics, but operate and describe the system under study differently. For example, density functional theory (DFT) is based on the electron density, while the Hartree-Fock method (HF) is based on the wave function. Quantum mechanics/molecular mechanics (QM/MM) is a hybrid method, aiming to combine the accuracy of quantum mechanics for the molecular regions under study with the efficiency of molecular mechanics (MM). The fragmented molecular orbital method (FMO) splits a large molecule into smaller fragments, with the aim of treating each fragment quantum mechanically and taking into account their interactions.
Many of the results obtained, with the integration of quantum computing, aim to accelerate the drug development cycle and improve workflows, model interactions between small molecules and macromolecules, understand the functioning of enzymes based on electronic effects, model protein-ligand interactions and much more. The increase in computational performance and the excellent results obtained identify the quantum mechanical approach as very promising in drug discovery. In the coming years, computational drug discovery combined with quantum mechanics is expected to become increasingly common.
References
- Sarfaraz K. Niazi. Quantum Mechanics in Drug Discovery: A Comprehensive Review of Methods, Applications, and Future Directions, Int. J. Mol. Sci., 2025.
- Yidong Zhou, Jintai Chen, Jinglei Cheng, Xu Cao, Yuanyuan Zhang, Gopal Karemore, Marinka Zitnik, Frederic T. Chong, Junyu Liu, Tianfan Fu, Zhiding Liang. Quantum-machine-assisted drug discovery, npj Drug Discovery, 2026.








